- 移动端


武汉康测科技有限公司
9 年
手机商铺
商家活跃:
产品热度:
- NaN
- 0.5
- 0.5
- 1.5
- 0.5
公司新闻/正文
LncRNA也能调控转录激活?
1449 人阅读发布时间:2022-11-25 14:42
研究背景
Linker组蛋白是一种高丰度的染色质相关结构蛋白,一般作为转录抑制因子存在。其中Linker组蛋白H1在核小体排列和折叠成更紧凑的染色质结构中起着至关重要的作用。可以阻止其他蛋白质与染色质的结合,还可以为转录激活因子或抑制因子提供招募平台来影响染色质功能。组蛋白在基因组分布不均匀,在染色质可及性区域高的位置富集较少,而在不可及染色质区域分布较多。
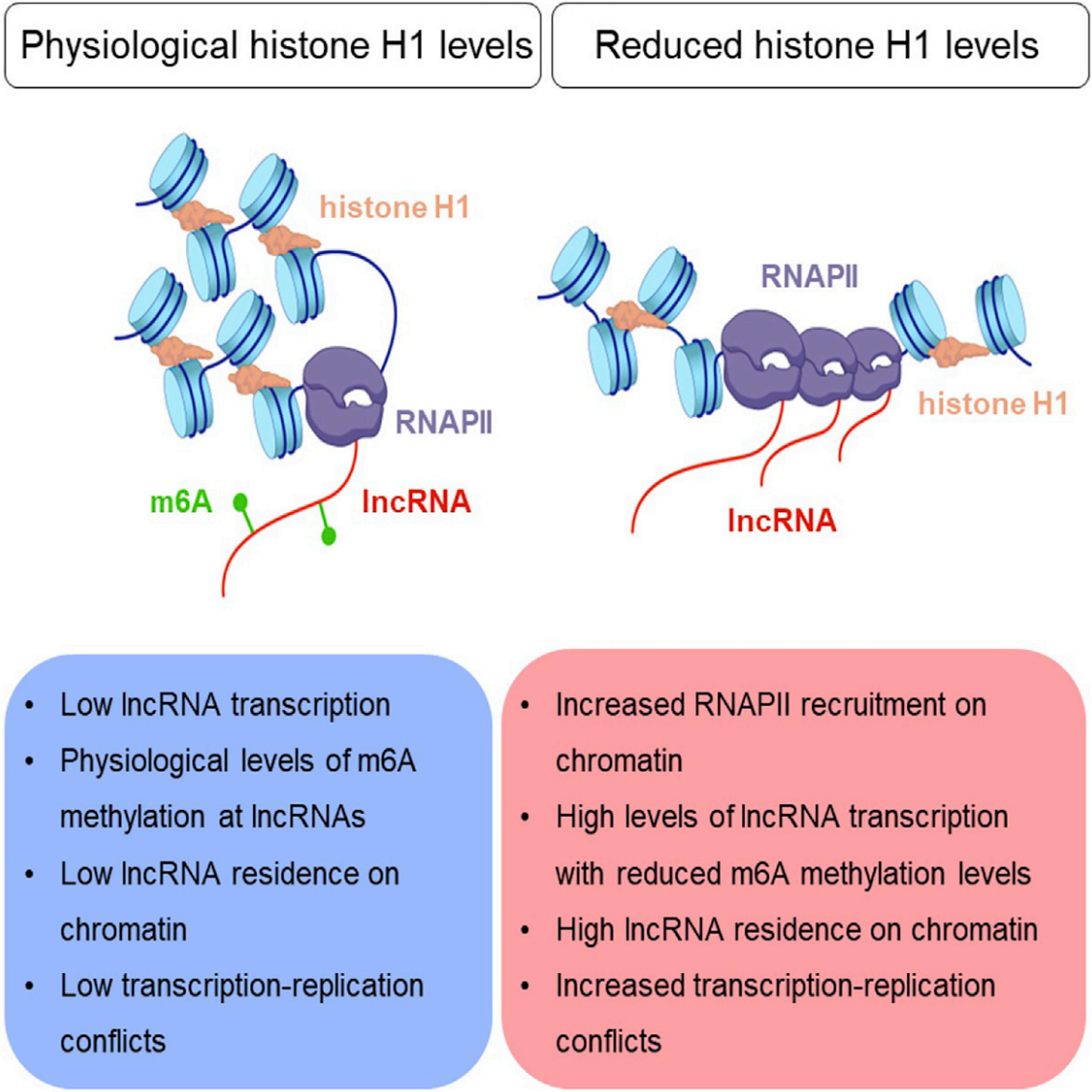
组蛋白H1含量的减少会导致复制起始模式的全基因组改变,以及由于复制-转录冲突而导致的复制叉停滞和DNA损伤。由此引发:H1缺陷后基因表达的有限改变如何与广泛的复制-转录冲突相协调?为了阐明组蛋白H1对转录调控的附加功能,作者对H1-tko缺陷的小鼠胚胎干细胞中RNA转录本丰度、RNA聚合酶II(RNAPII)的位置和活性以及新生RNA的m6A修饰分析,揭示了组蛋白H1在调节染色质中非编码RNA功能转换和m6A修饰的调控作用,让我们来看看作者是如何完成这项工作的吧。
研究结果
1.组蛋白H1含量的降低导致染色质上非编码转录本的积累
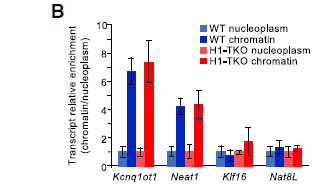
为了研究H1缺乏导致转录依赖性复制应激的机制,作者运用染色质富集非编码RNA测序(CheRNA-seq)技术检测H1-tko缺陷的小鼠胚胎干细胞富含染色质的RNA。通过检测两种与染色质转录后相关的非编码RNA(Kcnq1ot1和Neat1)相对于两种正常转运的mRNA(Klf16和Nat8L)的染色质/核质比值发现染色质富集非编码RNA中染色质相关转录本的富集程度更高。
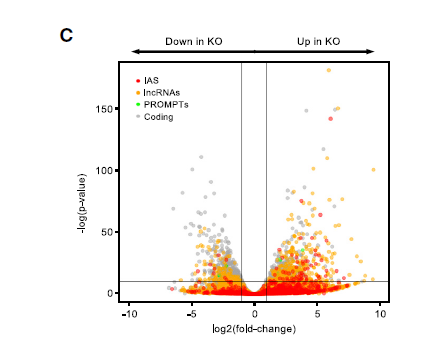
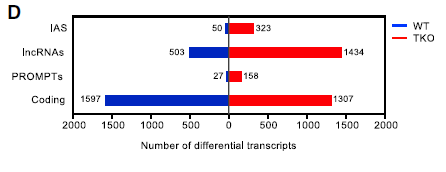
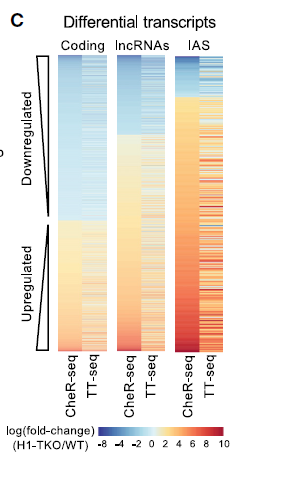
此外,分析显示差异表达的转录本包括四个类别根据其基因组位置和编码潜力分为四类:(1)内部反义RNA(IAS),(2)长基因间区非编码RNA(lncRNA),(3)启动子上游转录本(PROMPTs)和(4)编码RNA(mRNA)。值得注意的是,所有非编码RNA类别在H1-TKO细胞中都上调。这些发现表明,需要适当含量的组蛋白H1抑制染色质中非编码转录本的积累,如下图。
- 染色质累积的非编码RNA是调控型RNA
染色质相关的非编码RNA通常具有顺式调控功能。为评估组蛋白H1缺失后小鼠胚胎干细胞中的lncRNA是否增强了邻近启动子的活性,作者根据编码基因相对于其与差异表达的lncRNA的距离的表达水平计算得出编码基因越接近lncRNA,基因的转录活性越高(如图B)。并且许多上调的非编码转录本是从类增强子染色质区域产生的,且在H3K4me1的转录起始位点(TSS)周围显示富集,如下图C。
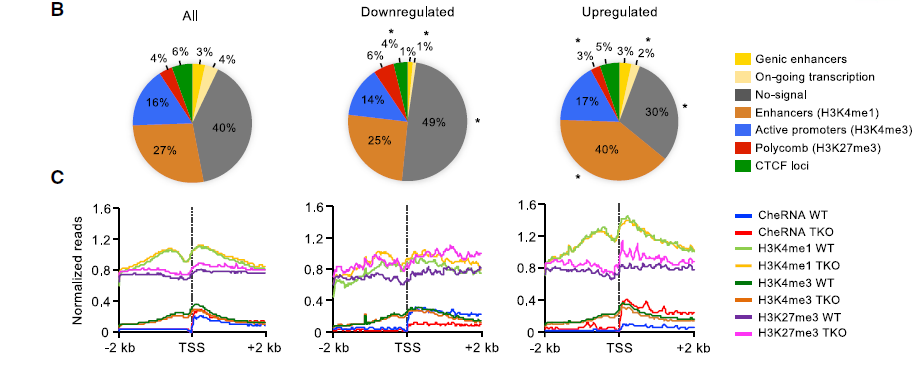
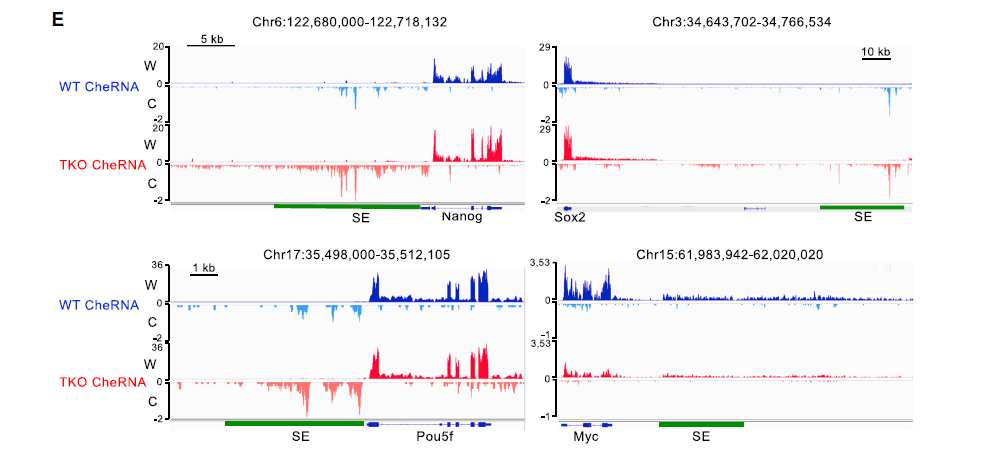
同时,在H1-TKO细胞中差异富集的lncRNA偏向位于参与发育和RNAPII转录的基因的顺式调控区域,这些基因是与细胞分化和多能性相关的转录因子(Nanog,Sox2,Pou5f等);这些lncRNA TSS定位在调控胚胎干细胞中这些基因转录的超级增强子(SE)区域,如下图E。
此外作者发现lncRNA表达与其相邻编码基因表达之间的差异倍数变化与下调相关,但与上调lncRNA无关,如下图。这些数据表明组蛋白H1是一种调控lncRNA的抑制因子,H1缺乏时,lncRNA在染色质中积累,可能有助于复制。
- 染色质累积的非编码RNA与转录依赖的复制应激有关
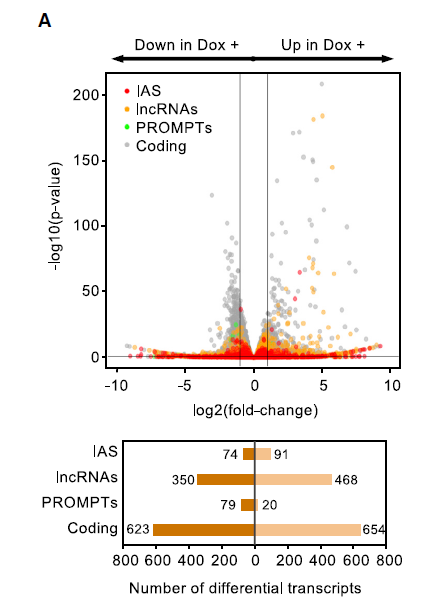
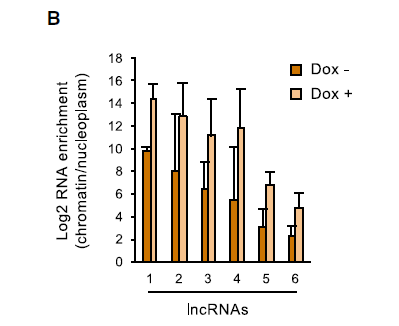
为了证实染色质中非编码转录本的积累是组蛋白H1缺失的结果,而不是与H1-TKO 小鼠胚胎干细胞缺乏分化潜力相关的间接效应,作者接下来分析了组蛋白H1敲低的人分化细胞的转录状态。应用此前的CheRNA-seq相同计算方式,重新分析了已发表的乳腺癌T47D细胞的RNA-seq数据,T47D细胞经在强力霉素诱导下敲低H1.2和H1.4亚型(shMultiH1,分别是鼠H1c和H1e的人类同系物)。尽管相对于CheRNA,非编码转录本在总RNA中的表达减少,但lncRNA转录在组蛋白H1沉默的诱导后增强(如下图A),且lncRNA在核质中富集(下图B深棕色),此外,在组蛋白H1耗竭后,差异lncRNA的在染色质中累积增加(如上图B浅棕色)。
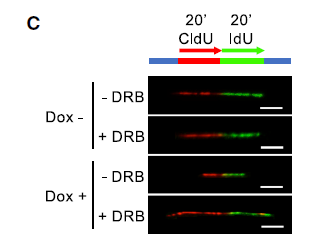
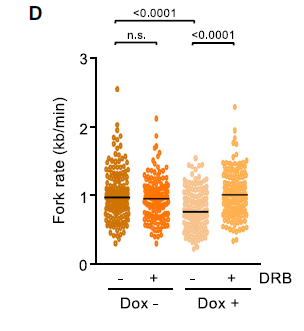
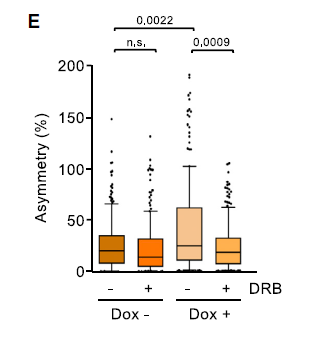
为了确认缺失组蛋白H1是否与DNA复制应激有关,DNA纤维分析显示,H1减少后复制叉出现率显著降低,复制叉不对称性增加(下图C-E);而且复制表型是转录依赖性的,因为当通过DRB处理抑制RNAPII延伸活性时,复制和转录活性都很容易恢复。这些细胞的DNA损伤信号传导升高,同时通过转录抑制而降低。总之,人类分化细胞中组蛋白H1含量的降低增加了转录依赖性复制应激,这可能是通过增强的非编码RNA染色质结合来介导的。
- 累积的非编码RNA通过RNAPII与染色质相连,可以形成R-loop
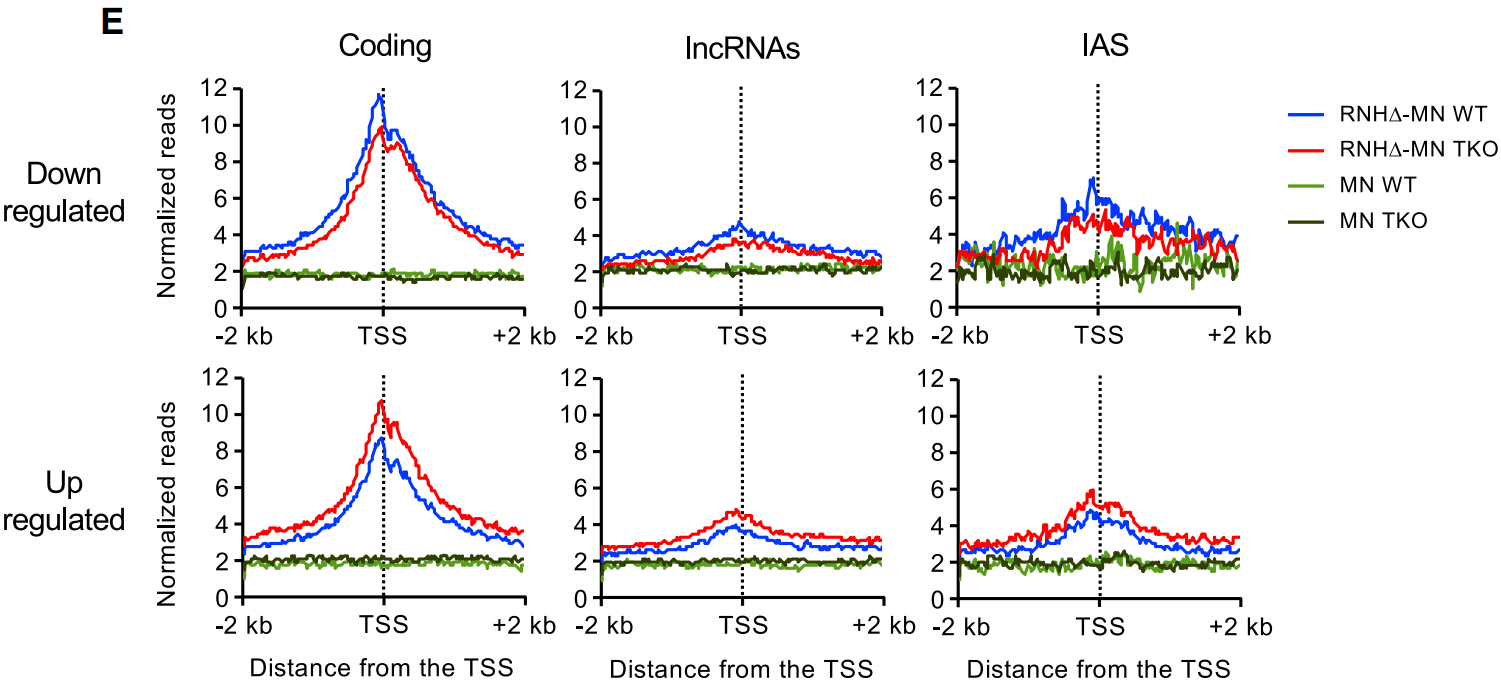
为了解决lncRNA在H1缺失时如何转录的问题,作者研究了RNAPII在基因组占位情况。首先使用人染色质作为spike-in对照进行染色质免疫沉淀测序(ChIP-seq),以检测细胞类型之间染色质结合RNAPII的定量差异,发现H1-TKO染色质中的RNAPII增加了12%,并且特异性地积累于TSS周围。不同细胞中lncRNA和IAS水平与编码RNA水平变化保持一致,在H1-TKO细胞中,上调的lncRNA和IAS的启动子近端区域招募的RNAPII几乎与一些编码RNA启动子一样多,如下图。
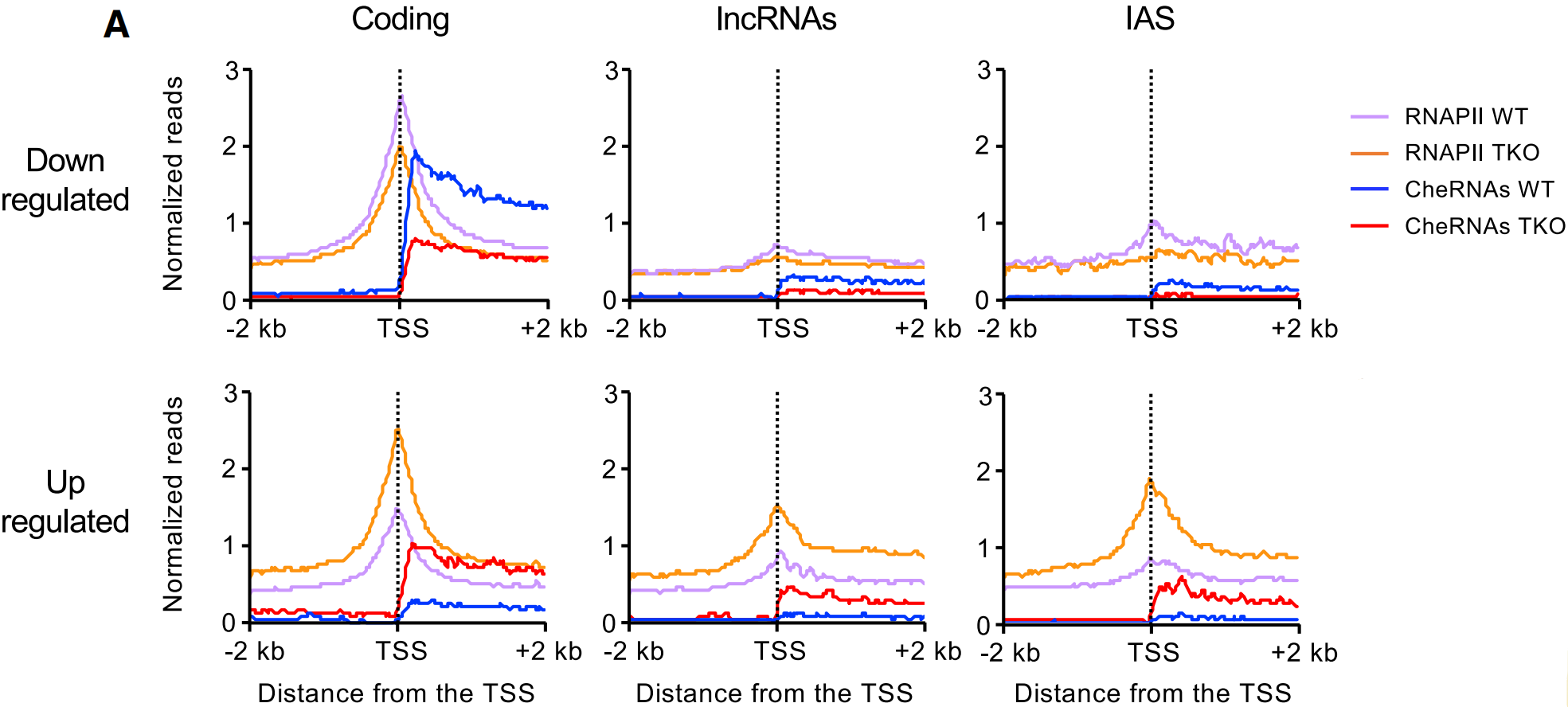
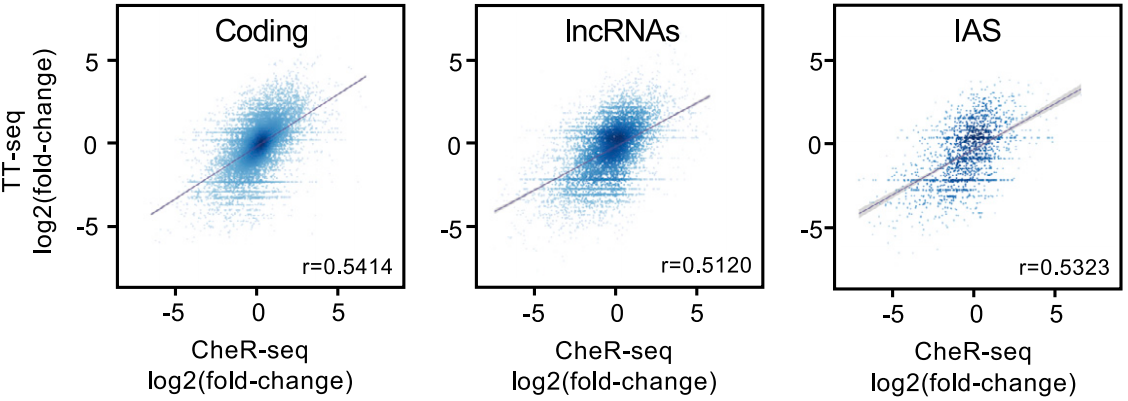
为了确保通过RNAPII复合物锚定在染色质上的差异转录本被顺利转录,作者还进行了新生转录组测序(TT-seq)。发现所有类别的RNA合成速率与染色质转录本丰度之间呈正相关,表明新生转录本在染色质上的停留时间与其产生有关,如下图。
在转录泡中停留时间较长的转录本可以产生RNA-RNA杂交结构(R环),这是哺乳动物细胞中复制-转录冲突的潜在来源。为了研究CheRNA位点的R环形成,作者通过MapR发现在所有转录本类别中,R环水平与CheRNA水平相似,证实组蛋白H1耗竭染色质中的R环形成增加,可能有助于复制表型。
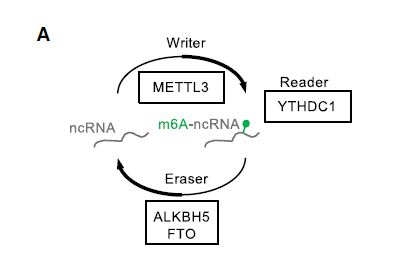
- 染色质相关转录本的m6A修饰水平降低
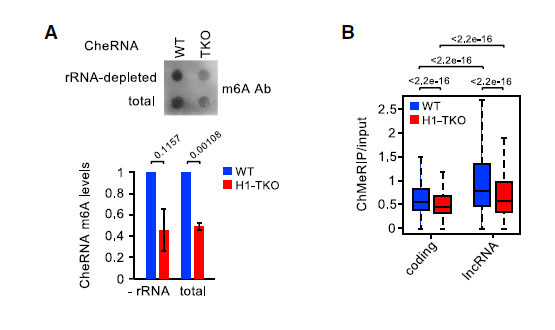
染色质相关的调控RNA(包括PROMPTs和增强子RNA)存在m6A修饰,这使它们在染色质中的水平不稳定。因此,为了鉴定被H1抑制的lncRNA转录物的特定特征,作者使用体外甲基化RNA作为spike-in对照,将CheRNA纯化后进行m6A meRIP-seq。分析显示,编码和非编码转录本的H1-TKO细胞中的m6A修饰水平显著降低,并且lncRNA在WT 细胞染色质中显示出比编码RNA更高的m6A水平,表明非编码转录本具有不同的转录后调控动态,如下图。
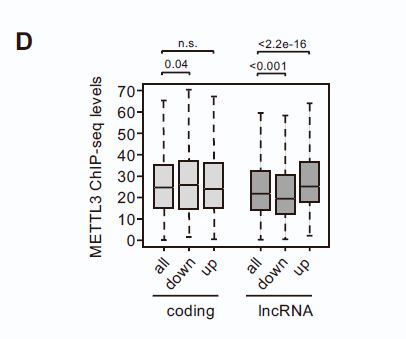
在H1-TKO细胞中,lncRNA中的m6A修饰显著降低,特别是表达水平上调的lncRNA。而在WT细胞中,其启动子区域的m6A Writer METTL3的结合密度始终较高,如下图。
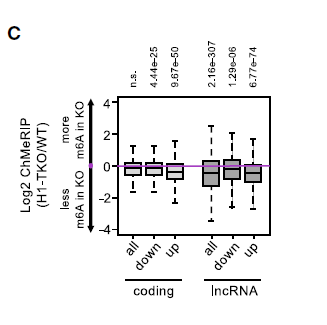
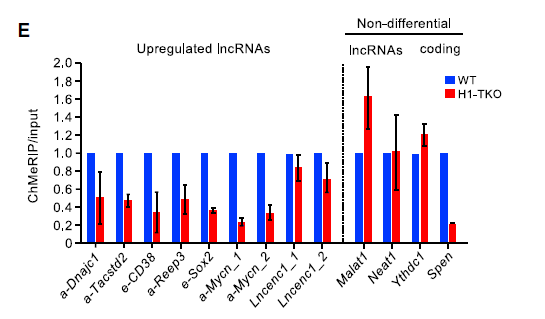
Ythdc1、Spen和lncRNA Malat1或Neat1转录本在H1-TKO染色质上富集程度没有差异;对m6A修饰丰度的定量评估显示在组蛋白H1缺失的细胞中m6A修饰水平增加或没有变化(编码转录阻遏因子Spen的mRNA在H1缺陷细胞中的甲基化显着降低),如下图。相比之下,所有上调的lncRNA在H1-TKO细胞中都显示出不同程度的m6A 修饰水平降低,表明m6A修饰降低是染色质积累lncRNA的标志。
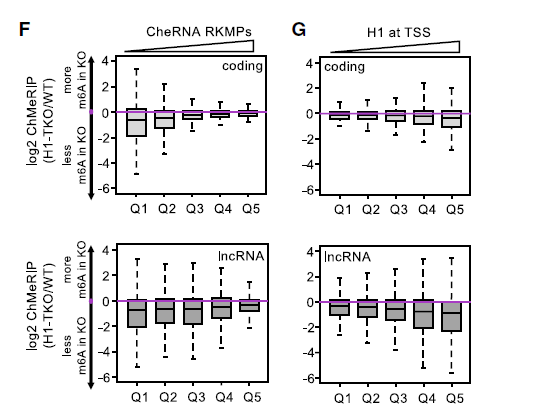
为表征主要受m6A缺失影响的转录本,作者根据WT细胞中新生的RNA丰度和组蛋白H1启动子-近端占据情况对其进行了分类,发现m6A水平的最大降低与WT细胞中较低的转录本表达和较高的组蛋白H1占有率相关。进一步证实了组蛋白H1和染色质相关lncRNA的m6A水平之间存在联系,如下图。
- lncRNA甲基化减少会改变其染色质转换,引发复制-转录冲突
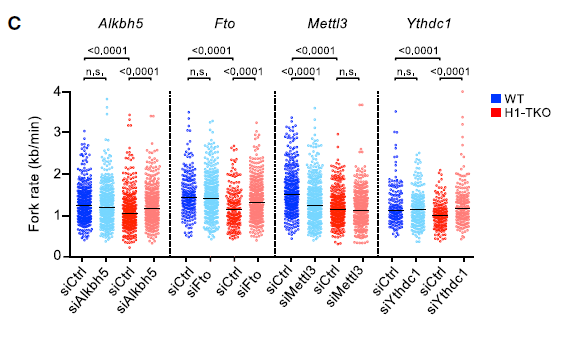
上述分析表明,组蛋白H1水平的降低导致染色质中非编码转录本以m6A依赖性方式改变。由此作者预测H1-TKO细胞中m6A修饰水平的增加将破坏染色质中非编码转录本的稳定性,从而减轻复制应激。为了验证这一预测,作者运用siRNA沉默m6A Eraser ALKBH5和FTO,分析纤维拉伸的复制动态。发现任何一种去甲基化酶的减少都可使H1-TKO细胞的复制叉恢复到WT水平。尽管染色质上有高水平的lncRNA,但复制表型的逆转仍会发生,这表明m6A修饰的相对减少是这些细胞复制损伤的主要原因,如下图。
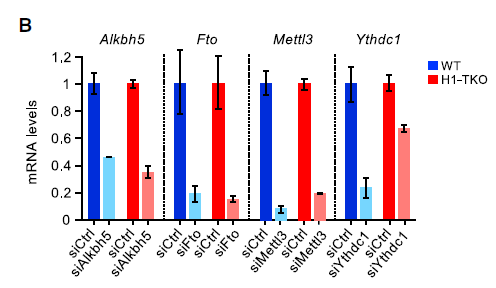
这一解释通过敲除m6A Writer METTL3得到证实,WT细胞中分叉缓慢延伸,而H1-TKO没有进一步复制。因此,METTL3似乎通过与组蛋白H1相同的通路调控染色质相关lncRNA上的m6A修饰水平。在敲除m6A Reader YTHDC1后,H1-TKO的复制叉也完全恢复,尽管它在H1-TKO细胞中的表达只降低了30%。总之,这些结果意味着需要适当的m6A修饰水平和reader YTHDC1在染色质结合的lncRNA上的作用,才允许平滑的复制叉前进过程,如下图。
研究总结
- 通过CheRNA-seq发现组蛋白H1缺失导致非编码RNA的转录并积累在染色质编码基因TSS区域;
- 通过分析顺式lncRNA作用距离,发现编码基因座位置越接近lncRNA,基因的转录活性越高;
- DNA纤维分析显示染色质累积的非编码RNA与转录依赖的复制应激有关;
- 通过RNAPII的ChIP-seq发现染色质积累的非编码RNA通过RNAPII与染色质相连,形成R-loop;
- 通过对染色质结合的非编码RNA进行m6A meRIP-seq,发现缺失组蛋白H1后这些非编码RNA的m6A修饰水平降低,通过敲低去甲基化酶可以减轻复制应激;
- 同样通过敲低reader蛋白YTHDC1也可以减轻复制应激;
引用文献
Fernandez-Justel et al., Histone H1 regulates non-coding RNA turnover on chromatin in a m6A-dependent manner, 2022, Cell Reports 40, 111329 September 13, 2022
DOI: 10.1016/j.celrep.2022.111329ll